The Procedure in Brief
Generally this technique involves the isolation of the RNA molecules from sources of interest and the separation of the isolated RNA by agarose or polyacrylamide gel electrophoresis, the separate RNA in the gel is then transferred to a solid matrix through the blotting process. The solid matrix, now with the separated RNA, is treated with labeled probe which specifically hybridizes or binds to complementary RNA sequence on the matrix. Unbounded probes are washed off the matrix. The labelled probe, now bound to complementary RNA sequence on the matrix are then read using a suitable detector. See fig 1 below(download the fig's picture and summary)
a: Electrophoresis of the RNA sample:
b:Transferring the separated RNA molecules to solid matrix (Blotting Process)
c: Blotting process continued
d: Exposing the memberane to labeled probes
e: The labeled probes hybridized to the RNA sample
f: The probes are detected with a suitable detector
Fig 1: A Pictorial outline showing a summarized process of Northern Blotting.
( source: http://teachline.ls.huji.ac.il/72320/methods-tutorial )
Extraction of RNA
The source of the RNA to be used for a northern blot analysis is wide and varied. It depends on the type of investigation that is being carried out. Some analysis such as, the characterization of the RNA constituents of retroviruses required that the viral RNA is degraded with specific ribonuclease before being subjected to electrophoresis.
A typical RNA molecule is extracted by homogenizing the tissue with a commercial reagent trizol (this aids in lysing the cell/tissue). After these initial lyses, the resultant mixture is extracted with concentrated solution of phenol (phenols disrupts the hydrogen bounding in the macro molecule causing proteins to derivative). The resultant mixture is centrifuged; a two phased supernatant is obtained. The upper phase contains the RNA and carbohydrate while the lower phase contains the DNA. The upper phase is collected and the RNA present is precipitated with alcohol. The resultant precipitate is further washed with alcohol.
For highly sensitive and specific assays, the RNA obtained in this way is further subjected to more purification schemes to obtain a highly pure RNA. Such scheme may include passing the RNA solution through a column of oligo(dT) probe which interacts with mRNA ployA tail. After extensive washing with the buffer, the mRNA is now eluted and then used for further analysis.
Electrophoretic separation of the RNA molecules
The RNA samples obtained after extraction contain many types of RNA molecule separation is essential since it can provide information on how many types of RNA is there and the estimated size. Electrophoresis is a technique used in separating biological molecules based on differences that exists in their sizes. By subjecting the samples to direct current, they acquire charge per mass ratio and then migrate through the agarose gel under the influence of the electric field. The rate of migration across the gel is function of the size and sometimes, a function of the shape of the molecule. Generally smaller molecules migrate faster through the gel.
The RNA samples (about 10-50mg) are treated with denaturating agents and heat before being subjected to the Electrophoretic separation. Common denaturants used for this purpose includes formaldehyde and glyoxal. The agarose gel to be used for the electrophoresis should also be prepared under complete denaturing condition. This may involve soaking the gel overnight in a solution of 1% SDS or other suitable commercial product. The denaturants should also be present in the anode and cathode buffer. This precaution about including a denaturing agent serves to prevent the purified RNA strands from forming base pair with themselves, it also serve to denature any ribonuclease that would interfere with the process.
During electrophoresis, the cathode and anode buffer should be continuously stirred to prevent a pH gradient from building up. Markers such as standard samples of ribosomal 18S and 28S RNA fraction. This two markers in addition to providing information on the molecular size of RNA molecules, also aids in monitoring of the blotting process, since they can detected by viewing the gel in a UV radiation at 254nm wavelength.
The Electrophoretic process may last for about 4-5hrs, the progress may be monitored by including a bromophenol blue- running dye along with the sample.
Blotting
This is the process of transferring the separated RNA molecules from the agarose gel to a suitable solid matrix. There are different kinds of blotting technique. The two most frequently used types are the capillary transfer and the electro blotting
.
Capillary transfer of the RNA molecules to a solid matrix involves the passive transfer of the RNA molecule unto matrixes like filter paper, nitrocellulose filter or nylon membrane under the influence of capillary force. The gel is placed on a sponge soaked in a buffer or salt solution. The nitrocellulose filter is placed on the gel slab and many layers of paper towels are placed on it (see fig1b). The solution essentially passes through the gel and nitrocellulose filter to the paper towel. This procedure may take about 12-16rs.
Electro blotting involves the use of electric current to transfer the RNA molecules from the gel to the solid matrix. Filter paper is the frequently used solid matrix. In this type of blotting, the filter paper is saturated with the transfer buffer. The gel is then sandwiched between the wet sheet of blotting paper and wet sponge. It is then rolled with a glass rod. This is now inserted into the blotting chamber. At this chamber, a voltage of 15v and about 0.21 ampere is allowed to flow across the blotting setup for about 12hrs.
The usual step after the completion of blotting is to ensure that the RNA molecules are properly bound to the solid matrix upon which it is adsorbed. This is done principally by exposing the membrane to UV. Exposure to UV causes the RNA molecule to undergo extensive cross linking. Another way of ensuring proper adsorption of the RNA molecules to the membrane especially if the membrane is to be stored for a longer period is by drying the membrane under vacuum.
After the blotting process, the next step in the northern blotting technique is the detection of the RNA molecule. The detection employs the hybridization principle.
Hybridization and Detection of RNA Molecules.
The principle of hybridization is based on the fact that two single strands of DNA will form base pair with each other so far they have similar nucleotide sequences that are complementary to each other. This base pair formation will occur regardless of the type of nucleic acid or the source from which it came from.
The application of the northern blotting technique requires that the researcher already knows the RNA of interest. This knowledge will aid in the design of a suitable detection probe. Detection probe essentially are labeled nucleotide sequence which are complementary sequence to the RNA being investigated the probe can be designed in different ways, the most important aspect of its design is getting a nucleotide sequence complementary to that being investigated. This could be obtained by getting the cDNA of the mRNA of interest either by chemical synthesis in the laboratory or by the use of appropriate plasmid.
Once the complementary nucleotide sequence is obtained, the sequence is then labeled.
There are two basic technique used in labeling the complementary nucleotide probe-The use of radioactive labeling and that of chemical labeling. The labeling of the probe aids in the visualization and quantification of the RNA of interest on the blotting membranes.
Radioactive labeling is usually done with 32P, inserted into the primer region of the sequence in the form of 32P-adCTP. Chemical labeling of the probe is usually done using enzymes which catalyses the breakdown of a chemiluminescent substrate, the reaction emits light that can be detected with appropriate device. Chemiuminesecent labeling can occur in a different ways. The nucleotide probe maybe attached to the enzyme directly or the ligand or antibody-labeled nucleotide probe may be attached to the enzyme. (See fig 2)
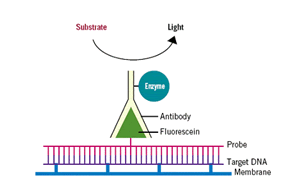 Fig 2: An outline showing a chemically labeled probe. In the presence of subtrate, the bound enzyme releases the fluorescein thus emitting detectable radiation. (Source: Osborn (2000)) Fig 2: An outline showing a chemically labeled probe. In the presence of subtrate, the bound enzyme releases the fluorescein thus emitting detectable radiation. (Source: Osborn (2000))
Procedure for hybridization and detection of the RNA
The blotted membrane, containing fixed RNA molecules are hybridized with the labeled probe by incubating the membrane in a solution containing the probe, this is usually done overnight in a rotating oven. During this process, the labeled probes complementary binds to any nucleotide sequence on the gel that has a nucleotide sequence complementary to it.
The excess and unhybridized labeled probe is washed off the membrane thoroughly; the hybridized membrane is then viewed with suitable detector based on the type of probe used. For radioactive probes, the membrane is detected with an autoradiography or a phosphoimager.
Chemical labeling requires that the membrane is incubated with appropriate reagent or substrate that would result in the emission of detectable light by the appropriate detector (fig 2). In both cases, observable dark bands are made on X-ray films exposed to the emitted radiation. (fig 3)
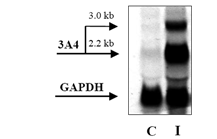 FIG 3: An example of Northern blot analysis. Northern blot of total RNA (20 μg) obtained from primary human hepatocytes using Trizol isolation procedure. C = control cells; I = cells treated with 3A4 inducer rifampicin. Band at 3.0 kb corresponds to 3A4 pseudogene. (Source :Dvořáka et al, 2003) FIG 3: An example of Northern blot analysis. Northern blot of total RNA (20 μg) obtained from primary human hepatocytes using Trizol isolation procedure. C = control cells; I = cells treated with 3A4 inducer rifampicin. Band at 3.0 kb corresponds to 3A4 pseudogene. (Source :Dvořáka et al, 2003)
The X-ray films obtained from the radioactive or chemiluminescent reaction can then be further analyzed with a computer densitometric software, that would quantitate the amount of RNA of interest that is present on the blotting membrane. |









